Group leader: Dr. Ruben van Boxtel
On the origin of cancer: studying somatic evolution in normal tissues.
 |
Identifying the rate limiting steps of cancer initiation in human tissues is challenging as many factors can play a role. The mutations in the genomes of cells can serve as an archive of their life history. We aim to decode these archives in order to pinpoint the initiation of cancer and identify causal processes in human tissues. To study the etiology of cancer, we have 3 research themes in our lab. |
Mutation accumulation in normal cells is required for the development of cancers. Because aging is the biggest risk factor for cancer, it is thought that mutation accumulation is rate limiting for disease initiation. Indeed, as our cells age, they gradually accumulate mutations in their genomes. If a cell by chance acquires a bad mutation at the wrong time and place, malignant transformation can be triggered. According to this model, we should target these mutations, or their functional effects, in order to remove the driving force of the cancer and kill it.
However, with the recent developments of sequencing normal, noncancerous cells, we see that the mutation loads in normal tissues are already so high that it is very likely that there are always cells in our bodies with the right set of mutations, which are required for malignant transformation; yet we don’t have cancer. For example, in quite some newborns we can detect leukemia-driving fusion genes in their blood, yet these babies are perfectly healthy and do not develop leukemia. In addition, for certain cancer types, such as leukemia, young children show a higher incidence compared to young adolescents. This phenomenon represents an apparent paradox, as young cells should have less somatic (oncogenic) mutations than adult cells. It is very likely that mutation accumulation, although very necessary, is not the rate limiting step in cancer development.
Why then, do certain individuals develop cancer and others don’t? If mutation accumulation is not rate limiting for disease development, what is? And more importantly, shouldn’t we be targeting those rate limiting factors? In other words, if getting cancer is default, because of the relatively high mutation burden in normal tissues, how do our bodies keep the initiation of cancer in check and why does this sometimes fail?
Pinpointing the rate limiting steps in cancer initiation is challenging as many factors can play a role: ecological features within the various tissues, tissue damage responses, age, carcinogenic exposure, etc. Still, an increased understanding in how the body can keep cancer in check might revolutionize how we battle the disease: we have “stolen” our best ideas from nature, so why not this?
To answer these questions, we have 3 research themes in our lab.
Theme 1: Tissue-specific mutation accumulation in human stem cells
Theme 2: Tracking the origin of cancer
Theme 3: The etiology of therapy-related malignancies in cancer survivors

Theme 1: Tissue-specific mutation accumulation in human stem cells
 |
Organ-specific cancer incidence varies significantly throughout the human body, which cannot be solely explained by different exposures to mutagenic environmental. Adult stem cells are likely the cellular targets for accumulation of pre-cancerous successive oncogenic hits, which eventually can give rise to tumor development, owing to their life-long capacity to propagate mutations to both self-renewing progeny and downstream progenitors. We aim to identify and study the mutational processes that are active in adult stem cells of various organs and precede oncogenic transformation. |
Organ-specific cancer incidence varies significantly throughout the human body, which cannot be solely explained by different exposures to mutagenic environmental factors, such as smoking, ultra-violet light or alcohol use. Adult stem cells are likely the cellular targets for accumulation of pre-cancerous successive oncogenic hits, which eventually can give rise to tumor development, owing to their life-long capacity to propagate mutations to both self-renewing progeny and downstream progenitors. We aim to identify and study the mutational processes that are active in adult stem cells of various organs and precede oncogenic transformation.
Strategy 1: Characterizing mutation accumulation during healthy life
Cataloguing rare somatic mutations in physiologically normal cells is technically challenging due to polyclonal nature of healthy tissues and the high error rate of single cell sequencing techniques. To overcome these challenges, we expand individual stem cells in vitro into clonal cultures, which reflect the genetic makeup of the original cell (i.e., mutations in the original cell will be shared by all other cells in the culture). Subsequently, sufficient DNA can be obtained to accurately catalogue the very rare somatic mutations present in the original cell. Using this approach, we have studied mutation accumulation in various tissues, such as blood, small intestine, colon and liver, and correlated this with organ-specific cancer incidence.
Publications:
Blokzijl et al., Nature 538:260-264 (2016) PMID: 27698416 Jager et al., Nature Protocols 13:59-78 (2018) PMID: 29215633 Osorio et al., Cell Reports 25:2308-2316.e4 (2018) PMID: 30485801
Rosendahl Huber A, Manders F, Oka R, van Boxtel R. Characterizing mutational load and clonal composition of human blood. J Vis Exp. 2019 Jul 11;(149)
Strategy 2: Study mutation accumulation in individuals at risk
Some individuals are at risk of developing cancer due to predisposing factors, such as germline mutations in DNA repair genes or a constitutive trisomy of chromosome 21 underlying Down syndrome. Indeed, newborns with Down syndrome have an increased risk of developing childhood leukemia, while they seem protected against solid tumors throughout life. We hypothesize that by characterizing mutation accumulation in stem cells of different tissues of individuals at risk for cancer, we will gain mechanistic insight into the rate limiting factors underlying carcinogenesis. Using this strategy, we found that fetal Down syndrome cells accumulate more mutations during early development as karyotypically normal cells. This increased mutation burden may contribute to the increased risk of leukemia early during life in Down syndrome.
Publications:
Hasaart et al., Scientific Reports 10:12991 (2020) PMID: 32737409 Hasaart*, Bertrums* et al., Expert Rev Mol Med. 23:e5 (2021) PMID: 33902785
Theme 2: Tracking the origin of cancer
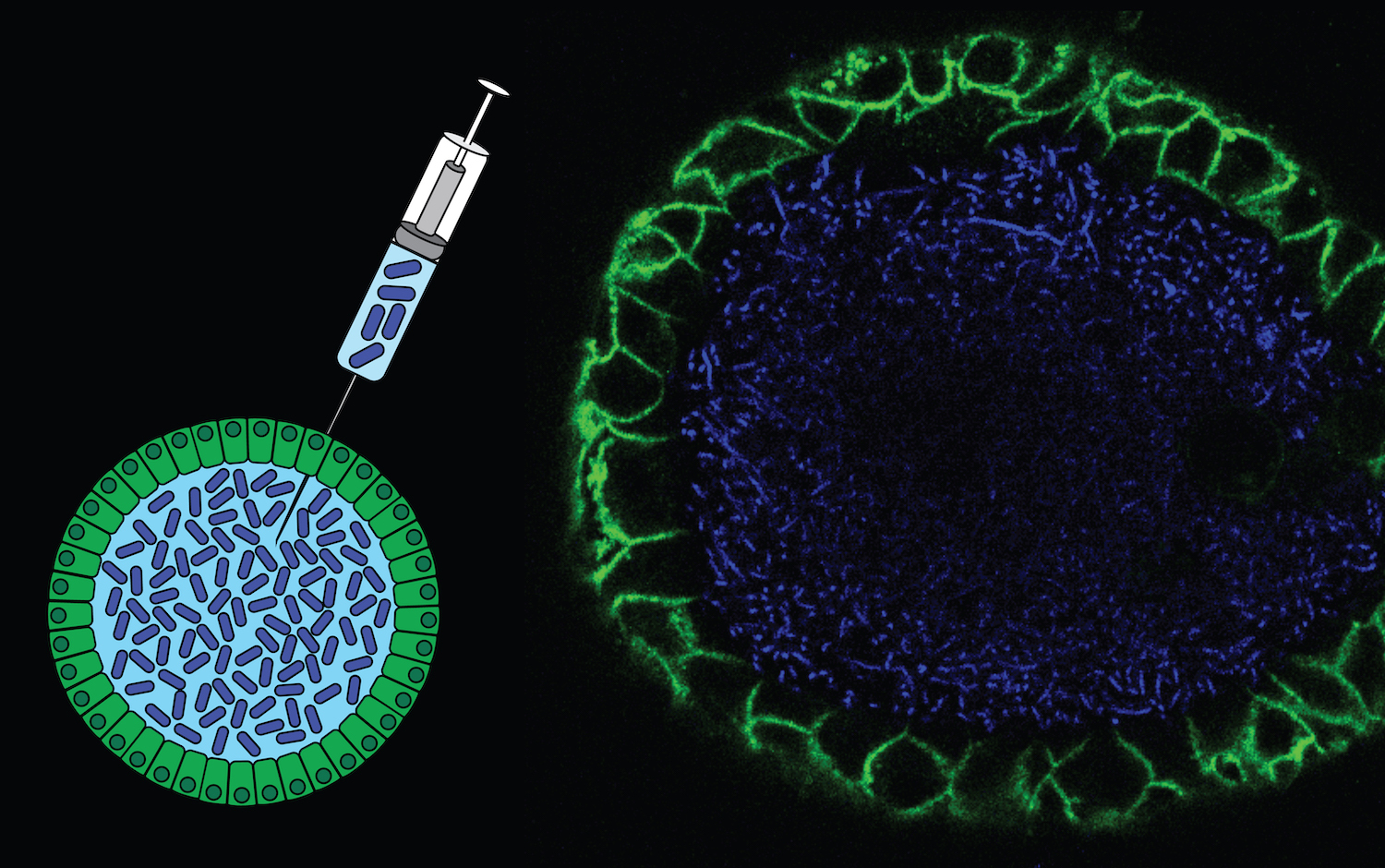 |
DNA is the largest biomolecule in the cells, which unlike other biomolecules is irreplaceable. The processes causing mutations leave characteristic patterns in the DNA, which can serve as a functional readout of mutagenic and/or DNA repair activity. In addition, phylogenetic relationships between different cells of the same individual can be exploited measure clonal dynamics within tissues. We aim to identify and study the mechanisms underlying characteristic mutation patterns in cancers as well as use mutations to retrospectively trace the cellular origin of cancer. |
Strategy 1: Dissecting the etiology of mutational signatures in cancer
A multitude of mutational signatures have been identified in cancer genomes. However, the mechanisms underlying many signatures remain unknown, which hampers their use in cancer diagnostics. We aim to functionally characterize the etiology of mutational signatures in cancer genomes. To achieve this, we combine experimental and bioinformatics approaches to systematically identify and characterize the processes underlying mutational signatures. For this, we apply CRISPR/Cas9 genome editing or specific carcinogenic exposure in human organoid cultures as well as cell lines. Using this strategy, we discovered a novel signature induced by common gut bacteria that can cause colorectal cancer. Also, we found a signature that can be predictive for genetic predisposition to cancer as a consequence of germline mutations in the DNA repair gene NTHL1.
Publications:
Drost et al., Science 358:234-238 (2017) PMID: 28912133 Blokzijl et al., Genome Medicine 10:33 (2018) PMID: 29695279
Pleguezuelos-Manzano et al., Nature 580:269-273 (2020) PMID: 32106218
Strategy 2: Retrospective lineage tracing of childhood leukemia and lymphoma
Why do children get cancer? For certain cancer types, such as leukemia, young children show a higher incidence compared to young adolescents. This phenomenon represents an apparent paradox, as young cells should have less somatic (oncogenic) mutations than adult cells. Therefore, the etiology of pediatric cancers may differ from cancer in the elderly. We aim to determine the rate-limiting steps underlying the genesis of childhood cancer by studying mutation accumulation and clonal lineages in blood of children with leukemia or lymphoma. For this, we study the processes that contribute to the development of childhood leukemia/lymphoma by comparing somatic mutation patterns between cancerous and patient-matched normal hematopoietic cells. Secondly, we aim to pinpoint the clonal origin of childhood leukemia during the development of the hematopoietic system by retrospective lineage tracing.
Publications:
Brandsma et al., Blood Cancer Discov DOI: 10.1158/2643-3230.BCD-21-0010
Theme 3: The etiology of therapy-related malignancies in cancer survivors
 |
Most chemotherapeutic drugs act by fatally damaging the DNA or blocking the replication thereof. However, noncancerous cells are also damaged by treatment, which can result in the accumulation of DNA mutations in normal tissues with potentially adverse effects later in life, such as novel malignancies. Our goal is to study the mutational effects of cancer treatment in normal tissues of children in order to develop novel treatment strategies aimed at minimizing or preventing adverse late effects. |
- Childhood cancer survivors are confronted with a variety of chronic health conditions as a consequence of their life saving therapy. Most chemotherapeutic drugs act by fatally damaging the DNA or blocking the replication thereof. However, noncancerous cells are also damaged by treatment, which can result in the accumulation of DNA mutations in normal tissues with potentially adverse effects later in life, such as novel malignancies. To minimize these adverse late effects, cancer treatment should be adapted to reduce mutagenicity for normal tissues while the cytotoxicity for cancer cells remains unaltered. However, the long-term mutational consequences of childhood cancer treatment in normal cells are currently unknown. Our goal is to study the mutational effects of chemotherapy in normal tissues of children in order to develop novel treatment strategies aimed at minimizing adverse late effects.
Strategy 1: Characterizing the mutational consequences of chemotherapy in normal cells of children
We systematically assess the mutational consequences of chemotherapy exposure in the hematopoietic system of children treated for cancer, as this tissue is extremely sensitive to chemotoxicity. For this, we compare the mutation burden of individual hematopoietic cells isolated of the same patient before and after exposure to chemotherapy and perform in-depth mutational analyses to pinpoint additive mutagenic effects of cancer treatment. We use the observed somatic mutations to examine the effects of treatment on the clonal composition and dynamics of the entire blood tissue. In addition, we have developed a unique cell screening assay to identify genotypes that increase the sensitivity of patients to the adverse mutagenic effects of chemotherapy. By systematically analyzing genome-wide mutational patterns resulting from such interaction, we aim to define genetic risk markers.
Strategy 2: Assessing the mutational consequences of hematopoietic stem cell transplantation
Genetic instability is a major safety concern for the use of stem cells in regenerative medicine. However, hematopoietic stem cell transplantation is routinely used for therapeutic purposes even though we do not know if this procedure can increase genetic instability and cause clonal hematopoiesis or even leukemia. Here, we address this question by comparing somatic mutations in the genomes of single HSCs before and after transplantation in multiple patients.
Strategy 3: Uncovering the rate limiting steps of therapy-related AML in childhood cancer survivors Therapy-related malignancies are a major cause of long-term mortality among childhood cancer survivors. However, it is unclear how exposure to chemo- and/or radiotherapy early in life induces carcinogenesis. Our aim is to determine the mechanisms and rate-limiting steps underlying the genesis of second malignancies in childhood cancer survivors. For this, we apply our unique methodology to study mutation accumulation at the single cell level in longitudinally collected blood samples of patients who developed of a therapy-related hematopoietic malignancy. Instead of focusing on identifying cancer driver mutations, we use the entire set of (mostly passenger) mutations in these cells to:
Track down the cellular origin of second malignancies by retrospective lineage tracing.
Identify the mechanisms causing second malignancies by in-depth mutational analyses.
Study phenotypic effects of cancer treatment on population dynamics of blood by integrating clonal histories and lineage contributions.
- 2025 - 2029 KWF Cancer Research Project Grant
- 2024 - 2029 VICI - News article
- 2024 - 2026 ERC Proof of Concept
- 2023 - 2027 Selected Oncode investigator Phase 2
- 2023 – 2028 NYSCF Robertson Stem Cell Investigator Award - News article
- 2023 – 2028 Ammodo Science Award for groundbreaking research (Omnes Pro Uno) - News article
- 2023 – 2026 Landsteiner Foundation for Blood Transfusion Research (LSBR) Research Grant
- 2022 – 2026 KiKa Research Project
- 2020 – 2023 KWF Cancer Research Project Grant
- 2020 – 2025 ERC Consolidator award (SecondCANCERinKIDS)
- 2019 – 2023 Selected Oncode Investigator after a highly competitive open recruitment call
- 2019 – 2023 KWF Cancer Research Project Grant
- 2018 – 2021 KWF Cancer Research Project Grant
- 2017 – 2022 NWO Vidi award
Poort VM, Hagelaar R, van Roosmalen MJ, Trabut L, Buijs-Gladdines JGCAM, van Wijk B, Meijerink J, van Boxtel R. Transient Differentation-State Plasticity Occurs During Acute Lymphoblastic Leukemia Initation. (2024) Cancer Res 84(16):2720–2733
Bertrums EJM, de Kanter JK, Derks LLM, Verheul M, Trabut L, van Roosmalen MJ, Halse H, Antoniou E, Reinhardt D, Dworzak MN, Muhlegger N, van den Heuvel-Eibrink MM , Zwaan CM, Goemans BF, van Boxtel R. Selective Pressure Of Platinum Compounds Shape The Evolution Of Therapy-Related Myeloid Neoplasms. (2024) Nature Comm 15:6025
Rosendahl Huber A*, Pleguezuelos-Manzano C*, Puschhof J*, Ubels J*, Boot C, Saftien A, Verheul M, Trabut LT, Groenen N, van Roosmalen M, Ouyang KS, Wood H, Quirke P, Meijer G, Cuppen E, Clevers H#, van Boxtel R#. Improved detection of colibactin-induced mutations by genotoxic E. coli in organoids and colorectal cancer. (2024) Cancer Cell 42(3):487-496.e6.
Middelkamp S, Manders F, Peci F, van Roosmalen MJ, González DM, Bertrums EJM, van der Werf I, Derks LLM, Groenen NM, Verheul M, Trabut L, Pleguezuelos-Manzano C, Brandsma AM, Antoniou E, Reinhardt D, Bierings M, Belderbos ME, van Boxtel R. Comprehensive single-cell genome analysis at nucleotide resolution using the PTA Analysis Toolbox. (2023) Cell Genom. 3:100389.
Bertrums EJM, Rosendahl Huber AKM, de Kanter JK, Brandsma AM, van Leeuwen AJCN, Verheul M, van den Heuvel-Eibrink MM, Oka R, van Roosmalen MJ, de Groot-Kruseman HA, Zwaan CM, Goemans BF, van Boxtel R. Elevated Mutational Age in Blood of Children Treated for Cancer Contributes to Therapy-Related Myeloid Neoplasms. (2022) Cancer Discovery 12:1860-1872.
de Kanter J, Peci F, Bertrums E, Rosendahl Huber A, van Leeuwen A, van Roosmalen MJ, Manders F, Verheul M, Oka R, Brandsma AM, Bierings M, Belderbos M#, van Boxtel R#. Antiviral treatment causes a unique mutational signature in cancers of transplantation recipients. (2021) Cell Stem Cell 28:1726-1739.e6.
Brandsma AM, Bertrums EJM, van Roosmalen MJ, Hofman DA, Oka R, Verheul M, Manders F, Ubels J, Belderbos ME, van Boxtel R. Mutation signatures of pediatric acute myeloid leukemia and normal blood progenitors associated with differential patient outcomes. (2021) Blood Cancer Discov 2 (5): 484–499
Peguezuelos-Manzano C*, Puschhof J*, Rosendahl Huber A*, van Hoeck A, Wood HM, Nomburg J, Gurjao C, Manders F, Dalmasso G, Stege PB, Paganelli FL, Geurts MH, Beumer J, Mizutani T, Miao Y, van der Linden R, van der Elst S; Genomics England Research Consortium, Garcia KC, Top J, Willems RJL, Giannakis M, Bonnet R, Quirke P, Meyerson M, Cuppen E, van Boxtel R#, Clevers H#. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. (2020) Nature580:269-273
Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, Nijman IJ, Martincorena I, Mokry M, Wiegerinck CL, Middendorp S, Sato T, Schwank G, Nieuwenhuis EE, Verstegen MM, van der Laan LJ, de Jonge J, IJzermans JN, Vries RG, van de Wetering M, Stratton MR, Clevers H, Cuppen E, van Boxtel R. Tissue-specific mutation accumulation in human adult stem cells during life. (2016) Nature 538:260-264. PubMed PMID: 27698416
*Equal contribution

















